
Written by Adam Hospital, IRB Barcelona
Molecular simulation techniques are becoming crucial in the multidisciplinary approach to fight CoVid-19 and thousands of groups around the world are running simulations on CoVid-19 systems. Many of these researchers are using high performance computer (HPC) centres, such as the ones that are part of the ICEI/Fenix Research Infrastructure. This article briefly introduces the work that the BioExcel Center of Excellence (BioExcel CoE) is doing on the CoVid-19 research, taking advantage of the resources granted through the PRACE-ICEI Calls for Proposals.
BioExcel CoE is the European central hub for biomolecular modelling and simulations. Started as an H2020 EU-funded project in 2015, it has now become a reference for the Life Science biomolecular simulation field. Partners involved in the project are the main developers of the most popular software tools in the field, including GROMACS (MD simulations), HADDOCK (Docking), pmx (free energy) and CP2K (QM, QM/MM).
Atomistic MD simulations were present in many of the BioExcel CoVid-19 related projects started during last year (2020). The first project, in collaboration between BioExcel and the Molecular Sciences Software Institute (MolSSI), was a community-driven data repository and curation service for molecular structures, models, therapeutics, and simulations related to CoVid-19 computational research: the CoVid-19 Molecular Structure and Therapeutics Hub. The repository was designed from scratch to share data from the scientific community, making science and data completely open to better tackle the CoVid-19 global emergency. The Hub has become a reference repository with huge amounts of useful information gathered in one single portal, with a particular focus on atomistic MD simulation generated trajectories.
These trajectory data is directly linked to the BioExcel-CV19 database and associated web server, which expands the power of the Hub, including interactive graphical representations of the MD trajectories and related pre-computed analyses. BioExcel-CV19 is a tool for scientists interested in the CoVid-19 research to interactively and graphically check key structural and flexibility features stemming from MDs, such as interface observables like residue distances (see figure below), hydrogen bonds and electrostatic interactions.
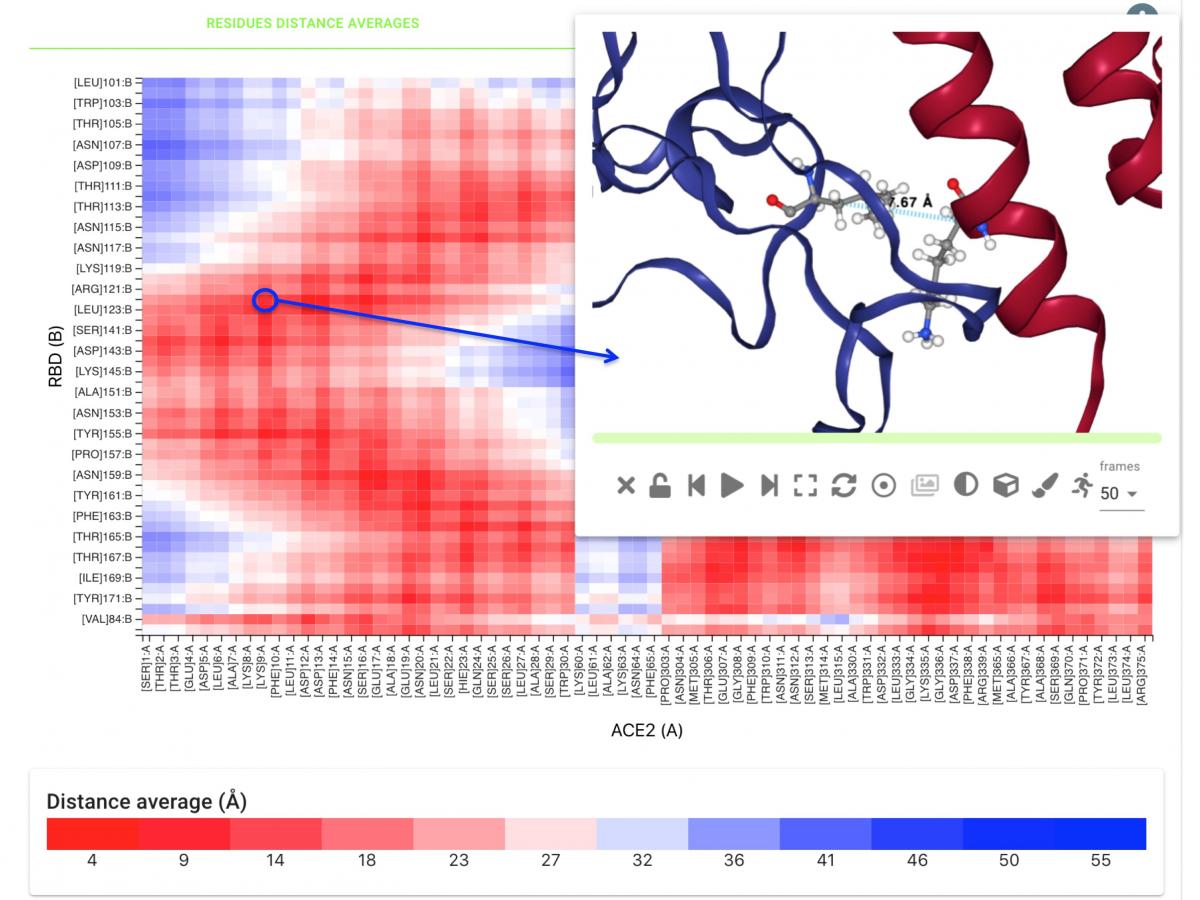
The Fenix resources granted through the PRACE-ICEI Calls for Proposals for the BioExcel CoE have allowed the calculation of atomistic molecular dynamics (MD) simulations of different SARS-COV-2 protein molecules, exposing insights on the effect of variants in the virus infectivity, both from the human cells point of view (polymorphisms) as well as from the virus point of view (variants, strains). MD trajectories computed in these resources have been uploaded into the BioExcel-CV19 database, including human Angiotensin Converting Enzyme 2 (hACE2), SARS-CoV-2 Receptor Binding Domain (RBD), RBD-hACE2 complex, and the SARS-CoV-2 Spike protein.
A combination of different BioExcel tools: GROMACS (MD package), pmx (free energy), BioExcel Building Blocks library (workflow development), and PyCOMPSs framework (workflow management), allowed an accurate prediction of the variant effects on the virus infectivity and the impact of the molecular flexibility, with at the same time making an efficient usage of the HPC computational resources. Results from the study includes dramatic differences in the interfaces of RDB and hACE2 in different species, recognition patterns across the different species, the possibility of an intra-species evolution (and associated zoonotic transfer), and the impact of the human polymorphisms and virus variants on the virus infectivity. Drafts for these projects are currently in preparation.
This work was partly done using the ICEI/Fenix Research Infrastructure resources. More information can be found in the 9th Fenix Infrastructure webinar given by Modesto Orozco (IRB Barcelona) and from the following links and references.
Useful Links:
BioExcel Center of Excellence (BioExcel CoE)
Molecular Sciences Software Institute (MolSSI)
CoVid-19 Molecular Structure and Therapeutics Hub
BioExcel-CV19 database and server
References:
Tejedor E, Becerra Y, Alomar G, et al. PyCOMPSs: Parallel computational workflows in Python. The International Journal of High Performance Computing Applications. 2017;31(1):66-82. doi: 10.1177/1094342015594678
Pau Andrio, Adam Hospital, Javier Conejero, Luis Jordá, Marc Del Pino, Laia Codo, Stian Soiland-Reyes, Carole Goble, Daniele Lezzi, Rosa M. Badia, Modesto Orozco & Josep Ll. Gelpi. BioExcel Building Blocks, a software library for interoperable biomolecular simulation workflows. Nature Scientific Data, 09/2019, Volume 6, Issue 1, p.169, (2019). doi: 10.1038/s41597-019-0177-4
Mark James Abraham, Teemu Murtola, Roland Schulz, Szilárd Páll, Jeremy C. Smith, Berk Hess, Erik Lindahl. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, Volumes 1–2, 2015, Pages 19-25, doi: 10.1016/j.softx.2015.06.001
Vytautas Gapsys, Servaas Michielssens, Daniel Seeliger, and Bert L. de Groot. pmx: Automated protein structure and topology generation for alchemical perturbations. J. Comput. Chem. 36:348-354 (2015). doi: 10.1002/jcc.23804
